Лютенурин
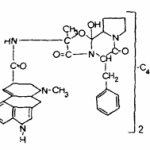
 Лютенурин представляет собой сумму дигидрохлоридов двух алкалоидов — нуфлеина (I) и тиобинуфаридина (II).
Лютенурин представляет собой сумму дигидрохлоридов двух алкалоидов — нуфлеина (I) и тиобинуфаридина (II).
Содержание суммы дигидрохлоридов в конечном продукте в пересчете на сухое вещество должно быть не менее 97 и не более 103%; содержание нуфлеина дигидрохлорида в сумме должно быть не менее 52%.
Лютенурин представляет собой аморфный порошок кремового цвета с желтоватым, иногда сероватым оттенком. Легко растворим в воде, 95% спирте, практически нерастворим в бензоле. Допускается опалесценция или мутность водного раствора.
Лютенурин является активным противотрихомонадным средством, оказывает также бактериостатическое действие в отношении грамположительных микробов и фунгистатическое действие на патогенные грибы типа Candida. Кроме того, лютенурин обладает сперматоцидной активностью. Применяют лютенурин для лечения острых и хронических трихомонадных урогенитальных заболеваний, трихомонадозов, осложненных бактериальной (грамположительной) и грибковой флорой, а также в качестве контрацептивного средства.
Формы выпуска: порошок для приготовления растворов, 0,5% линимент, вагинальные супозитории и пенообразующие таблетки, содержащие по 0,003 г препарата.
Сырьем для производства лютенурина служат корневища с корнями кубышки желтой (Nuphar luteae L.) семейства Кубышковых. Это водное корневищное растение с плавающими и подводными листьями.
Дико произрастает в европейской части, Закавказье, Казахстане, Сибири — в стоячих или медленно текучих водах до 5 м глубиной.
Промысловые заготовки ведутся на Украине, в Воронежской области, Краснодарском крае и других районах РСФСР и БССР.
Высушенные корневища содержат 0,4 % суммы алкалоидов.
Подготовка сырья и экстракция суммы алкалоидов. Корневища кубышки желтой измельчают на дисковой мельнице до получения частиц размером 2—3 мм. В экстрактор загружают кубышку желтую и при перемешивании приливают 10% раствор аммиака. Массу перемешивают в течение 30 мин до однородного смачивания. После перемешивания в экстрактор подают дихлорэтан. При включенной мешалке экстракцию ведут в течение 2 ч, при этом реакция среды должна быть щелочной.
Первый дихлорэтановый экстракт сливают в сборник, затем фильтруют через сукно на друк-фильтре. В экстрактор из сборника заливают дихлорэтан для проведения второй экстракции.
Вторую экстракцию проводят так же в течение 2 ч. Реакция среды должна быть щелочной. Второй дихлорэтановый экстракт сливают в сборник. Аналогично проводят третью, четвертую и пятую экстракции. Пятый дихлорэтановый экстракт не фильтруют, его используют для проведения первой экстракции следующей загрузки.
Обработка дихлорэтановых экстрактов серной кислотой. Дихлорэтановый экстракт после отстоя загружают в сульфатор, в него же приливают первую кислоту от обработки IV дихлорэтанового экстракта предыдущей загрузки. Реакционную массу перемешивают 10 мин, затем дают отстояться в течение 30 мин. Реакция среды должна быть кислая. Отстоявшийся дихлорэтан сливают в сборник дихлорэтана, а водный слой — I сернокислое извлечение — в сборник сульфата. Дихлорэтановый экстракт возвращают в сульфатор и приливают вторую кислоту от промывки четвертого дихлорэтанового экстракта предыдущей загрузки. Дважды промытый кислотой дихлорэтан используют для проведения экстракции алкалоидов из сырья. Обработку II дихлорэтанового экстракта ведут аналогично обработке I экстракта с той лишь разницей, что на первое сернокислое извлечение берут II сернокислое извлечение от обработки I экстракта, а на второе извлечение — 10% раствор свежей серной кислоты. Третий и четвертый экстракты обрабатывают подобно первому и второму. Первые сернокислые извлечения, полученные от обработки первых трех экстрактов с содержанием суммы алкалоидов 0,8—1%, выводят и передают на дальнейшую обработку. 1 и 2-ю кислоты, полученные от обработки IV экстракта, используют для экстракции следующей загрузки. После семикратного использования находящийся в схеме дихлорэтан регенерируют.
Получение технической суммы алкалоидов. Полученное сернокислое извлечение загружают в аппарат и охлаждают до температуры 5—7°. Затем в тот же аппарат из мерника небольшими порциями при непрерывном перемешивании приливают 25% раствор аммиака (pH 7,5—8,0). Выпавший осадок сразу отжимают на центрифуге и промывают с выкладкой холодной водой при температуре 10—15° и снова отжимают на центрифуге. Промытый и отжатый осадок сушат на воздухе в течение 20—24 ч.
Очистка технического основания и получение лютенурина фабриката. Порошок суммы алкалоидов загружают в аппарат и при перемешивании приливают этиловый эфир. Реакционная масса должна быть щелочной, в противном случае приливают раствор 25% аммиака.
Осадку дают отстояться в течение 30—35 мин и эфирное извлечение декантируют. Подобные операции повторяют 3—5 раз до тех пор, пока свежеприлитая порция эфира не будет содержать алкалоиды. Осадок после проведения эфирной экстракции не утилизируется. Полученные эфирные экстракты объединяют и сушат свежепрокаленным поташом. Затем экстракт фильтруют на нутч-фильтре и промывают этиловым эфиром. Эфирный экстракт упаривают до 1/10 первоначального объема. Кубовый остаток сливают в эмалированный аппарат и при перемешивании приливают пятикратный объем петролейного эфира. При этом выпадает хлопьевидный осадок, которому дают отстояться в течение 5—10 мин, после чего экстракт декантируют, а осадок промывают небольшим количеством петролейного эфира.
В раствор алкалоидов в петролейном эфире добавляют свежепрокаленный поташ и после 2—3-часового стояния отфильтровывают на нутч-фильтре и промывают петролейным эфиром. Раствор алкалоидов заливают в фарфоровую чашу и пропускают через него свежеполученный хлористый водород до кислой реакции. Выпавший осадок лютенурина отфильтровывают на нутч-фильтре, промывают сухим петролейным эфиром, протирают через капроновую сетку, раскладывают на противни и сушат на воздухе 2—3 суток до полного удаления растворителя, затем просеивают через капроновое сито.
Выход в производстве составляет 65%.